研发周期太长,创新药项目如何估值?【干货推荐】

作者:前瞻产业研究院 . 蒲翔
《中国上市药品目录集》中收录了在中国获批准上市的药品,目前共收录了1136种上市药品,其中进口原研药和通过一致性评价的药品(仿制药)合计888种,占比78%以上,是我国市场上最主要的药品类型。截止2020年2月,我国已经批准上市的创新药仅有21种,仅占上市药品数量的1.8%。

在成熟的医药市场中,仿制药占有极高的比例,以美国为例,仿制药的处方占有率接近90%,仿制药的普及可以大幅降低患者用药成本,对改善民生有重要意义;创新药则处在医药产业的前沿,为人类提供战胜疾病的利器,有至关重要的作用。2019年美国FDA批准创新药48个,相比之下,我国虽然拥有规模较大的医药市场,但创新药仍有较大的发展空间。

然而,创新药的发展不能一蹴而就,在国际上,创新药的临床前研究、临床试验和核准上市等每一个环节都有着严格的审核标准,从创新药立项到产品成功上市销售,平均周期一般达到8-10年。研发周期长、市场瞬息万变以及技术风险点不确定,极大地增加了创新药项目的投资风险,也为创新药项目的价值评估带来挑战。
一、为什么创新药项目研发周期长?
在药企开发新药之前,需要进行一系列的调研和研究工作,从市场、流行病学角度出发选择药物治疗的疾病类型,从知识产权、国家政策和企业自身条件角度出发确定开发的药物品种。随后,在创新药项目立项以后,需要经过临床前研究、临床试验和核准上市三个环节,如下图:
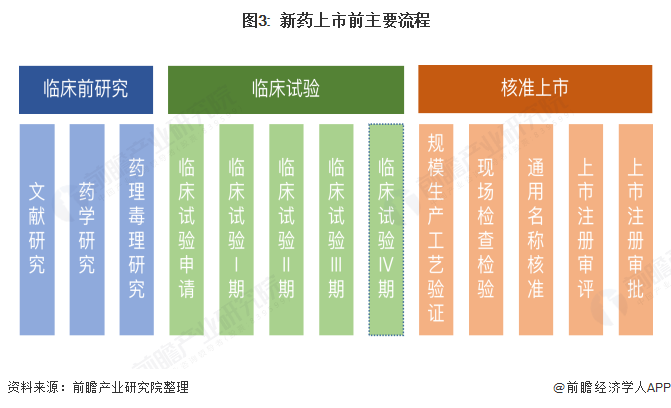
1.临床前研究
新药临床前研究阶段的内容包括文献研究、药学研究和药理毒理学研究。其中,药学研究又可细分为原料药的制备工艺、结构确证、制剂制备工艺、质量标准、检验方法、包材选择、稳定性研究等内容; 药理毒理学研究又细分为药效学、药动学研究,急性、长期及特殊毒理学研究等内容。
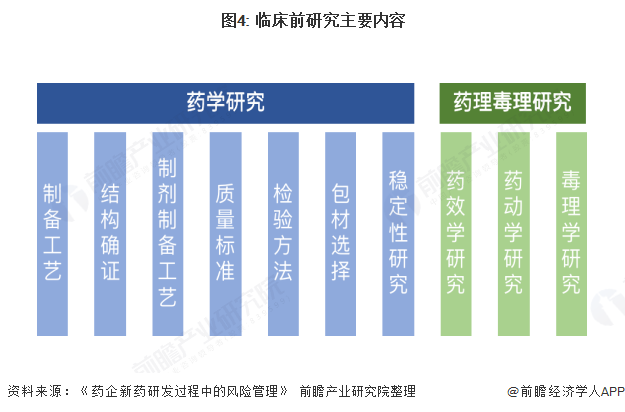
临床前研究一般需要12-24个月才能完成,研发周期长、过程复杂,在合成原料工艺与制剂处方等方面存在较高的技术不确定性,且稳定性实验比较耗费时间。
2.临床试验
药物临床试验申请:在创新药完成了药学研究和药理毒理研究等临床前研究后,向药品审评中心提出药物临床试验申请,中心接到药企申请后组织药学、医学和其他技术人员进行审评,并决定是否同意药企开展药物临床试验,该审批流程一般在60个工作日内完成。
在获得药物临床试验许可后,就可以进行后续的临床I、II、III和IV期临床试验,各期临床试验的主要目的如下:
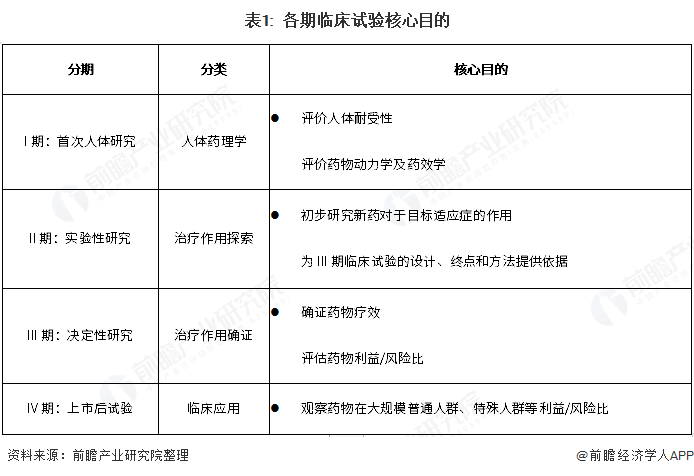
临床试验I期:在动物药理毒理试验基本成功的基础上,首次应用在人体上,用来初步评价新药的人体耐受性和药代动力学试验,主要研究药物的不良反应和药物被人体吸收代谢的机制。简单来说,临床I期试验主要研究药物的安全性。
临床试验II期:试验重点则转移到药效学上,主要目标是初步考察新药能否在目标患者人群中建立药效学相对比较一致、药物的毒性可以接受的水平,研究新药的血药浓度与药效学参数的关系,并为临床III期试验确定最大最小剂量范围。临床II期试验主要研究药物的有效性和给药剂量的关系。
临床试验III期:III期临床试验属于临床试验的治疗作用确证阶段,证明新药对目标适应症患者是安全有效的,其受益/风险比是可以接受的,为药物申报注册提供充分的依据,同时还为药品说明书和医生处方提供充分的数据。临床III期试验主要确证药物有效性,并评估其风险收益。III期试验完成后即可申请NDA注册上市。
临床试验IV期:新药上市后进行临床试验IV期,主要观察药物在广泛应用条件下的治疗作用和不良反应,并评价在普通或特殊人群中使用的收益与风险关系,并改进给药剂量。
从企业经营的角度看,创新药完成临床III期试验后即可申请注册上市,因此,更加关注临床I、II和III期的完成情况。
3.注册上市
根据我国药品注册管理办法,在完成支持药品上市注册的药学、药理毒理学和药物临床试验等相关研究,确定质量标准,并完成商业规模生产工艺验证和做好接受现场检查和检验的准备后,向国家局药品审评中心提出药品上市注册申请,中心组织药学、医学和其他技术人员在规定时限内对药品上市注册申请进行审评,一般在120个工作日内完成受理。
可见,我国新药产品在成功上市前需要经过复杂的研究、试验和审评。近年来,国家采取多项措施加速药物审评审批工作,境外上市新药在国内审评审批速度得到明显加速。对于创新药而言,审评审批周期占比较低,大量的时间主要投入在临床前研究与临床试验中,因此创新药从立项到上市的周期仍然较长。
二、创新药项目为什么难估值?
常规投资项目估值方法:对于投资类项目,一般通过企业自由现金流(FCFF)进行估值,即预测未来一定时期内项目能够产生的营业收入,并根据财务模型计算出相应的FCFF,并按加权平均资金成本(WACC)对FCFF进行折现并求和,得到当前时间点的财务净现值(NPV)。

用常规的FCFF估值方法对创新药项目进行估值,主要有2个问题:
1)预测周期长,估值误差大:创新药项目研发周期长,产生现金流入的时间点较晚,导致营业收入预测的误差较大,容易造成较大的估值误差。例如,某创新药项目在2020年初进行估值,但目前处于申请临床试验许可阶段,预计2028年才能实现上市销售,而市场形势瞬息万变,增加的这8年时间可能导致对营业收入的预测脱离实际造成较大误差;
2)无法体现关键节点估值变化:创新药项目的研发流程,有较为明显的标志性节点,例如获得临床试验许可、完成临床I期试验、完成临床II期试验等,从实际的案例来看,在完成每一个标志性节点后,创新药项目的价值都会有明显的跃迁,常规的估值方法无法体现创新药项目的标志性节点,并且没有考虑完成标志性节点后项目价值跃迁的情况。

因此,常规的FCFF估值方法无法直接用于创新药项目的估值,需要对其加以调整。我们以虚构的药物X为例进行演示。
三、创新药项目如何估值?
药物X是F公司正在研发的创新药,对小细胞肺癌有较好的治疗作用,每个患者平均需要5个疗程的治疗,每个疗程的治疗费用为1万元。目前X药已经获得临床试验许可,预计在2021年完成临床试验I期,2023年完成临床试验II期,2025年完成临床试验III期,2026年通过审批注册上市,2027年开始上市销售,专利在2036年到期。

我们先用常规的FCFF估值方法,计算项目在2027年初节点的估值。
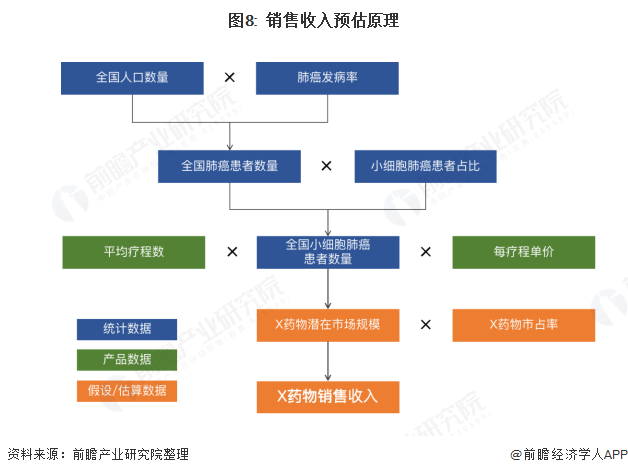
根据国家癌症中心发布的《2019年全国癌症报告》,2015年我国新发肺癌病例约为78.7万例,发病率为57.26/10万,其中小细胞肺癌占比约为15-20%,基于以上数据我们推算出每年X药物的潜在市场规模约为68.86亿元,在此基础上假设X药物的市占率即可推算出X药物的年销售收入。
按照创新药上市销售的销量增长来看,一般在上市销售10年左右达到销售峰值,随后由于专利保护到期仿制药进场导致单价和市占率快速下降。按照该规律,可以对X药物的上市销售后的销售曲线进行预估,如下图:
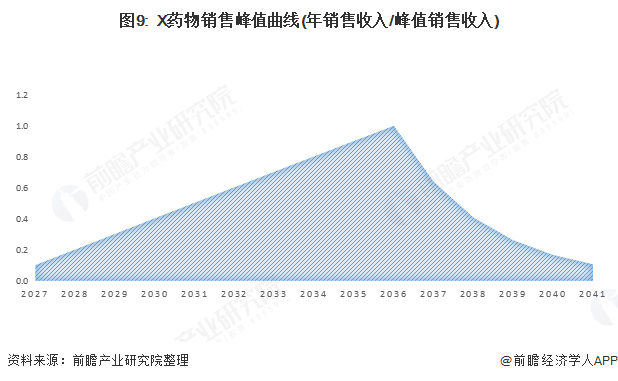
基于销售峰值曲线,我们进一步假设X产品峰值市占率达到10%,在2036年专利保护到期后由于仿制药入场售价和市占率快速下降,项目在2041年退出,推算出X药物在上市15年期间的销售收入,如下图:
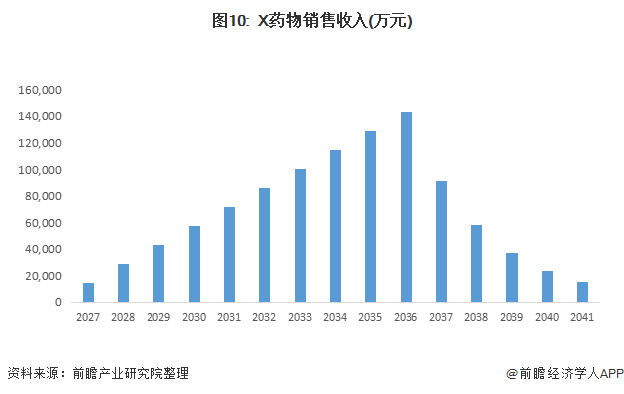
基于X药物的销售收入数据,我们结合财务模型计算出各年销售收入对应的企业自由现金流,并以6.5%的折现率计算X药物项目在2027年初节点的估值,为避免繁琐,此处省略计算过程,直接看结果。X药物项目在2027年初节点的估值PV6为18.6亿元。
截止目前,我们已经通过常规FCFF估值法计算出了X项目的估值,但这个方法引入一个问题,忽略了一个隐含的假设:X项目成功完成临床I期、临床II期、临床III期和审批注册上市的概率为100%。实际上,X药物项目可能在临床I期、II期等其中任何一个阶段由于安全性和有效性问题被放弃。例如,在进行临床I期时,发现X药物有较强的不良反应,需要额外进行巨额的研发费用加以改良,F公司评估收益后在此时放弃了X药物项目。因此,低估了X药物项目的风险,18.6亿元是虚高的估值。
为了解决这个问题,我们需要引入实物期权来修正估值方法。
由于国内已上市销售的创新药项目较少,各环节成功率的有效数据较少,我们参考国外医药市场的数据。Biomedtracker统计了2006-2015年期间的7455个新药研发项目,并估算了临床试验各阶段的成功率,如下:

我们基于临床试验各阶段成功率建立二叉树辅助估值,如下:
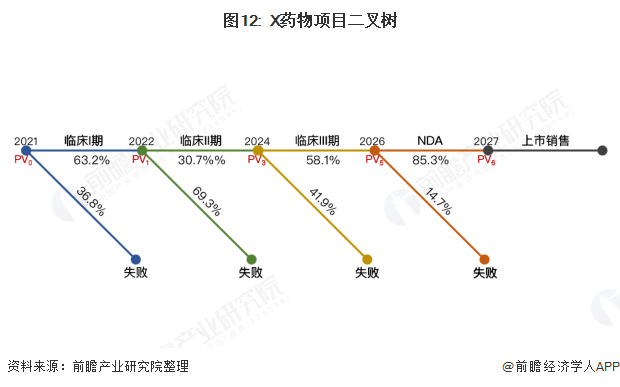
从二叉树可以看出,我们通过常规FCFF估值方法计算出的PV6是X药物项目成功通过前期所有阶段以后的估值。为了得到X药物在临床III期完成后的估值PV5,需要对PV6调整条件概率85.3%并折现(从2027年初折现至2026年初),如下:

以此类推,用同样的方法分别计算出PV3、PV1和PV0,即可得到X药物项目在各关键节点的估值,结果如下:
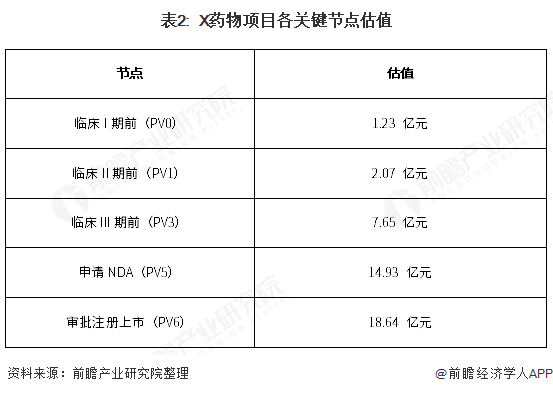
最后,由于X药物市占率、单价和财务模型中的关键比例等数据对项目估值结果有比较大的影响,且在估值过程中这些数据均取假设值,因此有必要对这些指标进行敏感性分析或情景分析,对不同情景下的项目估值进行比较,从而对创新药项目的投资价值做更好的判断。




















